
Alzheimer’s disease
Alzheimer’s disease is a progressive, incurable disorder of the brain, first described by Dr. Alois Alzheimer in 1906. In Alzheimer’s disease, brain regions critical for memory and understanding are slowly obliterated. This results in impairment of these functions, more commonly referred to as dementia. It is widely recognized that, while Alzheimer’s is the most common cause of dementia, it is actually a disease of the brain and is not a result of the normal aging process.
Approximately 24 million people worldwide suffer from Alzheimer’s disease, with age as the primary risk factor. It is estimated that up to half of people over the age of 85 suffer from Alzheimer’s. Because age is the greatest risk factor for developing Alzheimer’s, the upward trend in prolonged life span across the globe means that an ever-increasing number of aging people will be at risk of developing the disease. In addition, there are social and economic consequences to the growing prevalence of Alzheimer’s disease, as the stress on family members, carers, medical care providers and the State can be overwhelming. For these reasons, thousands of researchers across the globe are racing to find a cure for this devastating disease.
Alzheimer’s treatment and research
Current treatments for Alzheimer’s only address the symptoms of memory loss, but are unable to stop the advancing onslaught of brain deterioration. Over the last 100 years, huge advances in medical and scientific technology have vastly increased our understanding of the effect of Alzheimer’s disease on the brain. In addition to age, several genetic and environmental risk factors have been identified; however the true ‘smoking gun’ – a specific neurological event – has yet to be discovered. Thus, it is important not only to develop effective treatments for symptoms, but ultimately to identify the real cause and help successfully combat and prevent Alzheimer’s in the future.
Alzheimer’s disease is specific to humans and, because the brain cannot be safely biopsied, it is thus difficult to study the disease at early stages. However, over the last 15 years, scientists have been able to make use of genetic mutations to better comprehend the disease. These genetic mutations are the cause of Alzheimer’s disease in a small minority of patients (around five percent). This familial, or hereditary, form of the disease results from genetic mutations which may be passed down by either one or both parents. By inserting these mutated human disease-causing genes into mice, scientists have been able to create transgenic mice which develop Alzheimer’s when they normally would never do so. This allows researchers to look at Alzheimer’s disease in the brain as it develops over time, a feat impossible to do in the human brain. This work has had a significant impact on Alzheimer’s research and continues to shed light on the development of Alzheimer’s in the brain and improve our understanding of the disease, which will greatly accelerate the search for a cure.
Neurons relay information throughout the brain
The healthy brain contains around 100 billion neurons, the important cells which relay messages throughout the brain and are lost in Alzheimer’s disease. These neurons connect to each other at points called synapses and form a complex network of neuronal connections. In all, there are approximately 100 trillion synapses in your brain, where chemical and electrical signals are transmitted which allow you to move, think, learn, and interpret the world around you through sight, sound, touch, etc. Every bodily function and ability is controlled by the brain and, on a minute level, neurons.
The Alzheimer’s brain
The two main characteristic features, or pathologies, of Alzheimer’s disease are amyloid plaques, which are deposits of a small protein called amyloid-β (Aβ), along with neurofibrillary tangles, made up of clumps of an abnormal form of tau protein. Normally Aβ and tau function within neurons to control creation of neuronal connections, along with other tasks. However, abnormal forms of these proteins become sticky and cluster inside and between neurons. These aggregations of protein can then physically ‘get in the way’ of normal neuronal processes.
The large clumps of Aβ and tau occur specifically in regions of the brain designated for memory storage, learning, understanding, behaviour and personality. Ultimately, this leads to the destruction of synapses and neurons in these critical parts of the brain (Fig. 1). Essentially, Alzheimer’s attacks the very part of the brain that defines you as you.
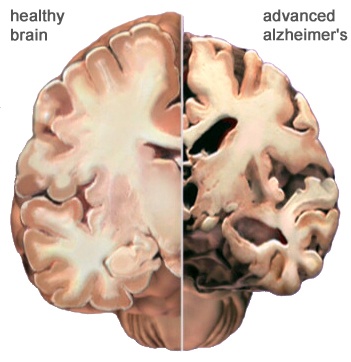
© 2007 Alzheimer's Association. www.alz.org. All rights reserved. Illustrations by Stacy Janis
Cellular communication in the brain
Messenger molecules
Within all cells, including neurons, there are hundreds of different proteins which act as ‘messenger molecules’ to transmit instructions or signals inside the cell and to surrounding cells. These messenger molecules have complex pathways – orders of signalling where messenger A ‘tells’ messenger B, which signals to messenger C, and so on, over many steps, in what is described as a signalling cascade, or pathway (Fig. 2). We can describe messenger A as ‘upstream’ of messenger B, and messenger C as ‘downstream’ of both A and B. Turning one messenger ‘on’ or ‘off’ can have several effects throughout the entire signalling cascade, meaning that the actions of only one messenger molecule can affect numerous neuronal functions.

Alzheimer’s disease and molecular ‘miscommunication’
Why do neurons die in Alzheimer’s disease? The answer to this question is not simple, as many factors come into play on a microscopic and molecular level, and we still do not know the primary causative factor in Alzheimer’s disease. Scientists have been able to study the pathology which builds up in the brain and causes neuronal death, but it is still unclear what initiates this accumulation. However, we do know that the process which makes neurons become ‘sick’ and die involves abnormal relaying of messages between neurons, and within the neuron itself.
The sticky proteins Aβ and tau clump together inside and outside neurons, preventing normal trafficking of messenger molecules inside neurons and across synapses. They are also able to turn essential messenger molecules ‘on’ or ‘off’, which can have drastic effects upstream and downstream in the signalling cascade. Research on brain tissue from patients who had Alzheimer’s disease has identified several messenger molecules which are defective in Alzheimer’s disease, but it is unclear if the resulting faulty communication is a cause or an effect of Alzheimer’s pathology.
The insulin signalling cascade
Insulin is a type of molecular messenger in the body called a hormone, and one of its functions is to regulate levels of sugar in the blood. It may be familiar to many readers as a treatment for diabetes. Insulin also has a key role in the brain, and is especially involved in memory and learning – the brain functions which are impaired in Alzheimer’s disease. Several studies, including work from our lab, have shown deficiencies in the insulin signalling pathway in the human Alzheimer’s disease brain. However, the exact relationship between insulin and the abnormal Aβ and tau remains obscure.
Results from my doctoral research
Transgenic animals have been instrumental in enhancing the understanding of Alzheimer’s, particularly a mouse named the ‘3xTg-AD’ model, in which Aβ and tau pathology closely mimic that found in the human brain. Thus, the focus of my doctoral work has been to utilize this mouse model of Alzheimer’s to examine certain molecular messengers in relation to the disease.
It was already known that insulin signalling was altered in Alzheimer’s, but my work set out to investigate if this defect was perhaps a cause of Alzheimer’s disease, or if it was a result of Aβ and/or tau pathology in the brain, and how these three relate to each other. One of the ways I did this was to use a technique called immunofluorescence, which allowed me to look at sections, or slices, of a mouse brain and ‘tag’ the messenger molecules with different colours. Using a microscope I then photographed the tagged proteins to examine where and at what levels they were present in the 3xTg-AD mouse brain in comparison to normal mice, and if they located near the clumps of sticky Aβ or tangled tau.
Insulin signalling is defective in transgenic Alzheimer’s mice
What I found throughout the course of my work is that the insulin receptor (IR), the protein to which insulin binds, is changed in the 3xTg-AD mouse. It is lost in the specific areas of the brain which have sticky Aβ and tau (Fig. 3a, b). The receptor also sticks to Aβ itself, and gets drawn into the large clusters of sticky Aβ known as amyloid plaques (Fig. 3c-e). What this means for the 3xTg-AD mouse is that there are fewer IRs to which the insulin hormone can bind. This has serious consequences for the affected neurons, as it means that other messenger molecules ‘downstream’ of the receptor may not be in their correct ‘on’ or ‘off’ state. This interferes with the ability of the neurons to carry out normal functions for survival. In addition, this deficit can impact the neurons’ ability to signal to other cells through damage to the synapses, the important connections between neurons, and ultimately results in neuronal death. Loss of synapses and the neurons themselves are the cause of impaired memory and other symptoms of Alzheimer’s disease.
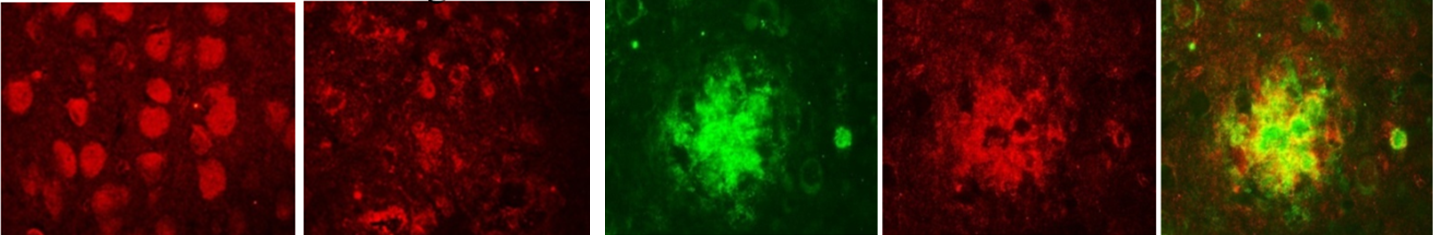
Insulin receptor (red) is found in neurons of a normal mouse (a), but is decreased in mice with Alzheimer’s pathology (b).
Clusters of sticky Aβ (green, c) draw insulin receptor (red, d) out of the neurons. By merging these images (e), it is possible
to see that Aβ and insulin receptor occur in the same place (yellow), providing evidence of a direct relationship between
the two proteins.
My work has also addressed a similar receptor called insulin-like growth factor-1 receptor (IGF-1R), which, as its name suggests, signals in a similar way to IR. IGF-1R is also changed in the 3xTg-AD mice, along with several other important messenger molecules downstream of both receptors.
Interestingly, defects in IR and IGF-1R associated more with Aβ, rather than tau pathology. This has allowed me to narrow the focus to possible interactions between Aβ and these receptors and the messenger molecules downstream in the pathways. What’s more, I have been able to extend my research beyond the 3xTg-AD mouse and look at neurons grown in the laboratory. These neurons were treated with Aβ and I was able to observe changes in IR and IGF-1R to provide further evidence of a relationship between Aβ and these signalling cascades in neurons.
Conclusion
The conclusion of this work is that over time, accumulation of Aβ in Alzheimer’s mice negatively impacts IR and IGF-1R signalling and their target proteins. Malfunction of these signalling pathways then has far-reaching consequences within neurons and how they communicate with other cells. Thus I have shown that the defects in several of the messenger molecules are actually a result, not a cause, of the disease, providing more information about how Aβ and tau affect the brain, an understanding which is essential to finding a cure. This work is the first to show such defects in an animal model of Alzheimer’s, supporting the hypothesis that insulin signalling plays a major role in Alzheimer’s, and should be taken into consideration when exploring new treatments.
Thanks to my supervisor Dr. Cora O’Neill, my colleagues in the lab and our collaborators, Dr. Dominic Walsh, University College Dublin and Prof. Frank LaFerla, University of California.Meghan Coakley is a student in the Neurobiology and Alzheimer’s Disease Laboratory under the supervision of Dr. Cora O’Neill. The author would like to acknowledge funding from the Science Foundation Ireland Research Frontiers Programme and the Health Research Board. All animals used in this study were treated humanely and in accordance with guidelines issued by the Irish Department of Health and Children.
